
16Sguild: Guild-based 16S-rRNA sequencing analysis pipeline
A Nextflow workflow for guild-based 16S-rRNA sequencing analysis such as introduced by Wu and Zhao et al., 2021.

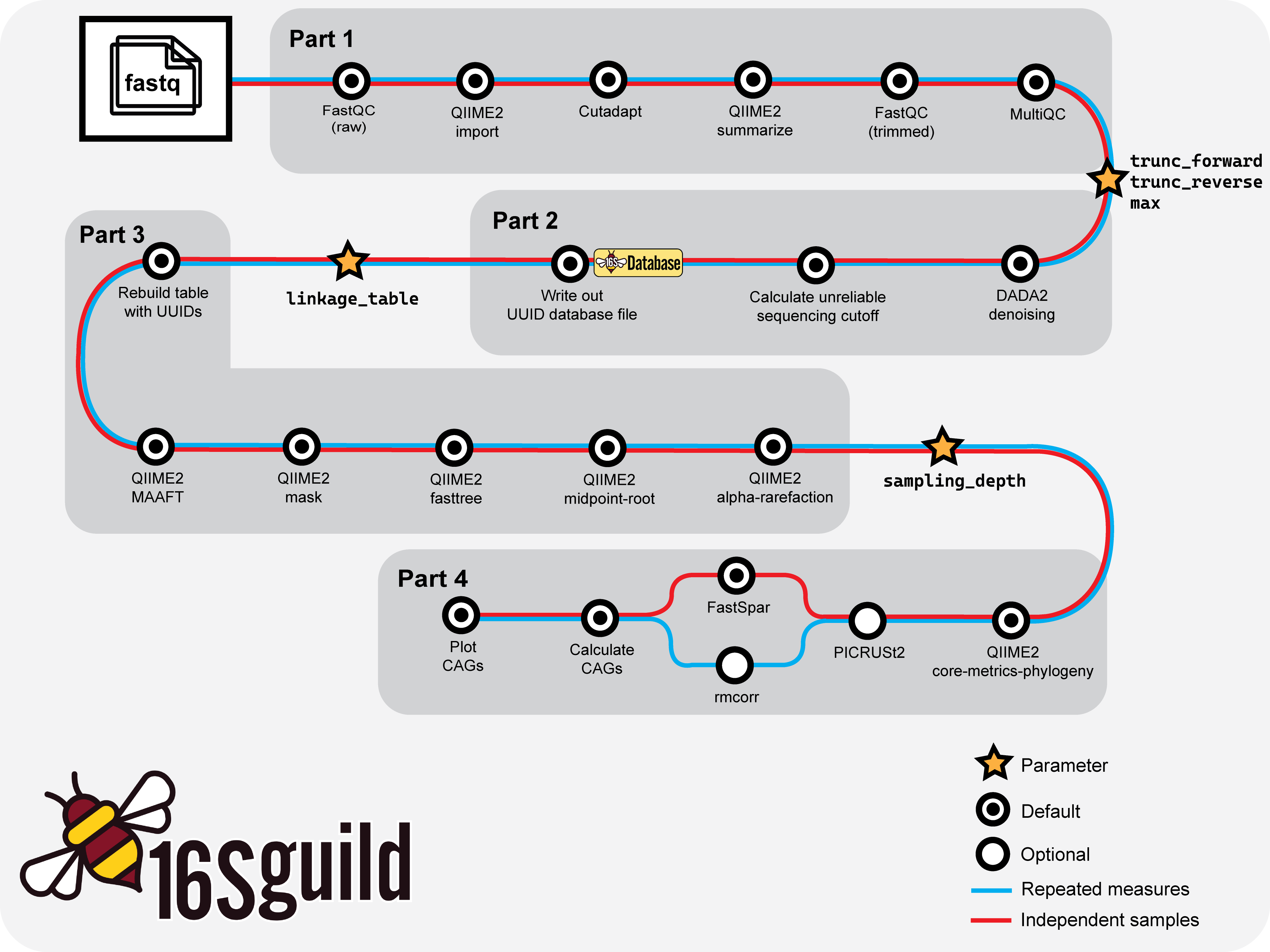
Basic workflow overview of 16Sguild pipeline.
Quick Start
Run directly from GitHub (suggested):
nextflow run zhao-microbiome-lab/16Sguild -params-file examples/params.yml
Or, clone the repository and launch the pipeline:
git clone https://github.com/zhao-microbiome-lab/16Sguild.git
cd 16Sguild
nextflow run main.nf -params-file examples/params.yml
Detailed usage examples can be found in the Basic Workflow documentation.
Note: The pipeline requires Nextflow (version 25.04 through 26.04) and either Docker or Singularity/Apptainer. See the Installation guide for details.
Parameter Configuration
Parameters are supplied in a config file, e.g. params.yml. Here is an example of a completed params.yml file. At the end of each part, users will need to examine their data and fill in the required values before continuing with their analysis.
# Required initial parameters
samplesheet: "samplesheet.csv"
metadata: "metadata.tsv"
base_name: "base_name_dataset"
input_type: "SampleData[PairedEndSequencesWithQuality]"
input_format: "PairedEndFastqManifestPhred33"
trim_forward: "GTGCCAGCMGCCGCGGTAA"
trim_reverse: "GGACTACHVGGGTWTCTAAT"
# Required parameters updated based on data
# End of Part 1
trunc_forward: "xxx"
trunc_reverse: "xxx"
max: "xxx"
# End of Part 2
linkage_table: /path/to/file.txt
# End of Part 3
sampling_depth: "xxx"
An example parameters file is available in examples/params.yml.
Brief Parameter Descriptions
| Parameter | Description |
|---|---|
samplesheet |
Path to the CSV file containing sample information |
base_name |
String identifying the name of input folder |
input_type |
Semantic type required for the QIIME2 import function |
input_format |
Format of the data to import (e.g., "PairedEndFastqManifestPhred33") |
trim_forward |
Forward primer sequence to trim |
trim_reverse |
Reverse primer sequence to trim |
metadata |
Path to the sample metadata table (TSV format) |
trunc_forward |
Position at which to truncate forward reads in QIIME2 DADA2 (experiment-dependent) |
trunc_reverse |
Position at which to truncate reverse reads in QIIME2 DADA2 (experiment-dependent) |
linkage_table |
Table from 16Sguild database assigning UUIDs to ASVs (experiment-dependent) |
max |
Maximum depth value used for alpha rarefaction (experiment-dependent) |
sampling_depth |
Rarefaction sampling depth for diversity analysis (experiment-dependent) |
Project Structure
main.nf– Main Nextflow pipelineprocesses/– Modular pipeline processes16SguildR/– Custom R package for downstream analysisbin/– Helper scripts (ensure they are executable)lib/– Definition script for end of part outputdocs/– Documentation for16Sguildpipelineconf/– Config profiles (e.g., for HPC, Docker, Singularity)examples/– Example input and config filesdocker/– Definition files for Singularity images automatically used in pipeline
Containers
This pipeline uses the following containers by default (defined in conf/default.config):
- FastQC:
docker://biocontainers/fastqc:v0.11.9_cv8 - MultiQC:
docker://multiqc/multiqc:latest - QIIME2:
docker://rachelgriffard/16sguild:qiime2_2024.2_1.0.0 - FastSpar:
quay.io/biocontainers/fastspar:1.0.0--h7f8d780_0 - R:
docker://rachelgriffard/16sguild:r_env_1.0.9 - Picrust2:
quay.io/biocontainers/picrust2:2.6.2--pyhdfd78af_1
No manual installation of software is needed—these containers are pulled automatically.
Output
- All results are written to the
results/directory by default. results/main_resultscontains output files from the pipeline.results/databasecontains the.rdsfile of the linkage table that can be uploaded to the database.results/visualizationscontains files that can be used on the QIIME2 viewing platform.- Intermediate files are preserved in the Nextflow work directory for reproducibility and in
results/intermediate_resultsfor easy access. - Results files are symlinked by default. Users can set
publish.mode = 'copy'to change this.
Testing
Test the pipeline with example data:
nextflow run main.nf -profile test
The test profile runs a minimal dataset and can be used for continuous integration.
Support
- For questions or issues, please open an issue.
- For feature requests, submit a new issue with the
enhancementlabel.
Citation
If you use this workflow, please cite: - This repository: zhao-microbiome-lab/16Sguild - Guild analysis: Zhao, Wu, & Zhao, 2024 - Nextflow: Di Tommaso et al. 2017
License
Distributed under the MIT License. See LICENSE for details.